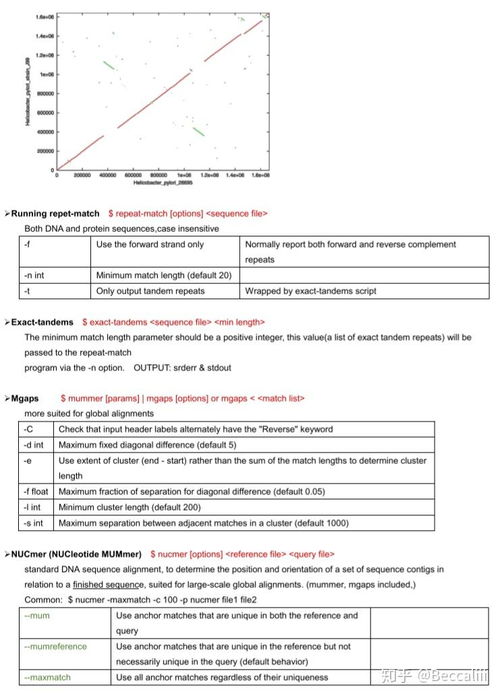
在生物信息学的世界里,序列比对是每个研究者都绕不开的一环。对于刚刚踏入这个领域的小伙伴来说,面对各种复杂的工具和软件,难免会感到一头雾水。今天,小明就来给大家分享他最近学习的一款超实用的序列比对工具——MUMmer。
### 什么是MUMmer?
MUMmer是一款专门用于DNA或蛋白质序列比对的强大工具,它可以帮助我们快速找到两条或多条序列之间的相似性和差异性。作为一名新手,小明一开始也觉得这款软件有些复杂,但经过一段时间的学习后,他发现其实MUMmer的操作并没有想象中那么难。
#### 安装与配置
首先,我们需要下载并安装MUMmer。小明选择的是Linux系统环境,因此他直接通过命令行完成了安装:wget https://downloads.sourceforge.net/project/mummer/mummer/3.23/MUMmer3.23.tar.gz。
接下来解压文件:tar -xvzf MUMmer3.23.tar.gz,然后进入解压后的目录:cd MUMmer3.23,最后编译安装:make。
#### 基础命令入门
安装完成后,小明迫不及待地开始尝试使用MUMmer进行序列比对。他首先准备了两段DNA序列作为测试数据,分别命名为ref.fasta和query.fasta。接下来,他运行了以下命令:nucmer --prefix=test ref.fasta query.fasta。
这条命令的作用是将参考序列ref.fasta和查询序列query.fasta进行比对,并生成一个名为test.delta的结果文件。
为了更直观地查看比对结果,小明还使用了show-coords命令:show-coords -rcl test.delta。这条命令可以将比对结果以表格的形式展示出来,包括序列间的匹配区域、长度、相似度等详细信息。
#### 实战演练
掌握了基本操作后,小明决定用真实的实验数据来检验一下MUMmer的能力。他从NCBI数据库下载了一组细菌基因组序列,并将其作为参考序列。接着,他又从另一项研究中获取了一些未知菌株的测序数据作为查询序列。
运行完比对后,小明惊讶地发现,MUMmer不仅能够准确识别出这些未知菌株与已知细菌之间的同源区域,还能高效处理大规模的数据集。这让他深刻体会到MUMmer的强大功能。
#### 小结
通过这次学习,小明总结出了几个关键点:
1. MUMmer是一款高效的序列比对工具,特别适合处理大型基因组数据;
2. 安装和使用过程相对简单,只需掌握几个核心命令即可上手;
3. 结合其他工具(如show-coords),可以进一步提升分析效率。
如果你也想了解MUMmer的更多功能,不妨亲自尝试一下吧!相信你一定会爱上这款强大的生物信息学利器。

发表评论 取消回复