在生物信息学领域,数据分析是一项既复杂又充满挑战的任务。今天,我将带大家走进一个强大的工具——maftools包,它可以帮助我们轻松地分析突变数据,并绘制出精美的瀑布图。
作为一名初学者,我在刚开始接触这个领域时也感到十分迷茫。但是,当我深入了解了maftools包后,一切变得简单起来。首先,我们需要了解什么是突变数据。突变数据是指基因组中发生的变异信息,这些信息对于研究癌症等疾病具有重要意义。
那么,maftools包是如何帮助我们分析这些数据的呢?maftools包是一款基于R语言开发的工具,它提供了丰富的功能来处理和可视化突变数据。接下来,我将通过实际操作向大家展示如何使用maftools包进行数据分析。
第一步:安装与加载maftools包
在R环境中,我们可以使用以下命令来安装maftools包:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("maftools")安装完成后,我们需要加载maftools包:
library(maftools)第二步:准备输入文件
maftools包支持多种格式的输入文件,其中最常用的是MAF(Mutation Annotation Format)文件。这种文件包含了详细的突变信息,如基因名称、突变类型、样本ID等。为了方便大家理解,这里提供一个简单的示例文件结构:
Hugo_Symbol Tumor_Sample_Barcode Variant_Classification
TP53 Sample1 Missense_Mutation
BRAF Sample2 Nonsense_Mutation第三步:读取并分析数据
使用read.maf函数可以轻松读取MAF文件,并将其转换为可用于分析的对象:
mobj <- read.maf(maf = "example.maf")接下来,我们可以调用各种函数对数据进行深入分析。例如,summarymaf函数可以生成一份详尽的数据概览报告,帮助我们快速了解数据的整体情况。
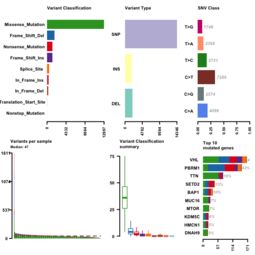
第四步:绘制瀑布图
瀑布图是一种直观展示突变数据的方式,它以矩阵的形式呈现每个样本中的突变情况。maftools包中的oncoplot函数可以帮助我们轻松绘制出这样的图表:
oncoplot(mobj, top = 20)以上代码将会生成一张包含前20个高频突变基因的瀑布图。通过这张图,我们可以清晰地看到哪些基因在不同样本中发生了突变。
总的来说,maftools包为我们提供了一种高效且直观的方法来分析突变数据。即使是没有太多编程经验的小白,也可以通过学习这篇教程快速上手。希望大家能够在自己的研究中充分利用这一工具,探索更多有趣的现象。




发表评论 取消回复