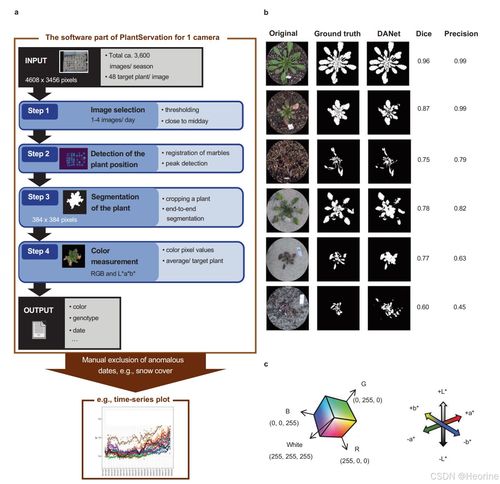
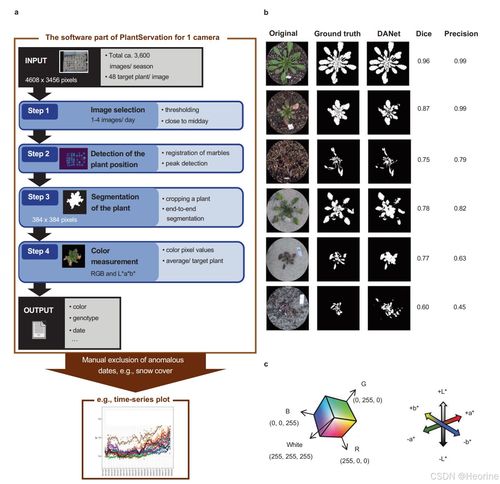
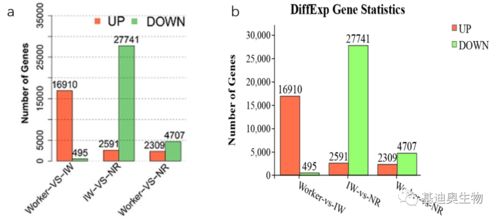
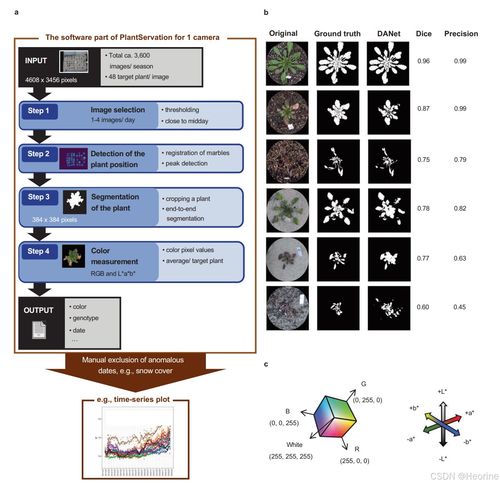
在科研的道路上,数据分析和可视化是每个研究者不可或缺的一部分。今天,我将分享一段特别有意义的经历——如何利用R语言中的ggmsa包来展示多序列比对(MSA)的结果,并结合Nature Ecology & Evolution期刊上的研究案例,让数据变得更加直观且易于理解。
为什么选择ggmsa包?
作为一名生物信息学爱好者,我在处理多序列比对时,常常需要一种简单而强大的工具来呈现复杂的序列差异。ggmsa包正是这样一个利器。它基于ggplot2框架开发,允许用户以高度自定义的方式绘制多序列比对图。无论是学术论文还是报告演示,ggmsa都能满足需求。
Nature Ecology & Evolution的研究背景
让我们先回顾一下2024年10月发表于Nature Ecology & Evolution的一篇重要文章。这项由中国科学院上海免疫与感染研究所Daniel Falush研究组主导的研究,揭示了一种古老的幽门螺杆菌生态物种。通过分析不同菌株的基因组数据,研究人员发现了其独特的进化特征。这一发现不仅加深了我们对幽门螺杆菌多样性的理解,还为后续相关疾病的研究提供了新视角。
类似的研究中,科学家们通常会使用多序列比对技术来比较不同样本间的遗传变异。然而,如何将这些复杂的比对结果清晰地展示出来,成为了一个挑战。这正是ggmsa大显身手的地方。
ggmsa的实际应用
以下是我的实际操作过程:
- 首先,确保你的R环境中已经安装并加载了ggmsa包。如果尚未安装,可以通过
install.packages("ggmsa")命令完成安装。 - 接下来,准备你的多序列比对文件。通常情况下,这类文件是以FASTA格式存储的。你可以通过读取函数将其导入到R环境中。
- 然后,调用ggmsa的核心绘图函数,例如
ggmsa(),并传入你的序列数据。此外,还可以根据需要设置颜色方案、字体大小等参数,使图表更加美观。
下面是一个简单的代码示例:
library(ggmsa) # 加载ggmsa包
seq_data <- read.fasta("alignment.fasta") # 读取多序列比对文件
ggmsa(seq_data, color_scheme = "clustal", font_size = 8)优化图表效果
为了让图表更具吸引力,我还尝试了一些高级技巧:
- 添加注释:通过额外的文本层或形状标记,突出显示关键区域。
- 调整布局:合理安排坐标轴标签、图例位置等元素,确保整体布局紧凑而不拥挤。
- 导出高质量图像:最终生成的图表可以保存为PDF或PNG格式,方便插入到文档中。
总结与展望
通过这次实践,我深刻体会到ggmsa包的强大功能及其在科学研究中的广泛应用。它不仅简化了多序列比对结果的可视化流程,还激发了我对数据呈现方式的新思考。未来,我计划进一步探索其他类似的工具,不断提升自己的技能水平。
如果你也对生物信息学感兴趣,不妨试试ggmsa吧!相信它会让你的数据故事更加精彩。

发表评论 取消回复