大家好,我是小李,一名生物信息学爱好者。今天想和大家分享一下我在学习WGCNA(Weighted Gene Co-expression Network Analysis)过程中的一些心得和经验。最近在简书平台上看到了很多关于WGCNA的文章,但总觉得缺少一些实际操作的细节。于是,我决定把自己这段时间的学习成果整理出来,希望能帮助更多像我一样的初学者。
首先,让我们简单回顾一下WGCNA的基本概念。WGCNA是一种用于构建加权基因共表达网络的方法,广泛应用于基因表达数据的分析中。通过这种方法,我们可以发现基因之间的相关性,并进一步探索这些基因在网络中的模块化结构。这对于理解复杂疾病的分子机制具有重要意义。
接下来,我们进入今天的重点——WGCNA的全流程代码分享。为了让大家更好地理解和应用这些代码,我会尽量详细地解释每一步的操作,并提供一些实用的小技巧。
1. 数据准备
在开始分析之前,我们需要准备好基因表达数据。通常,这些数据可以从公共数据库如GEO、ArrayExpress等下载。下载后的数据格式可能是原始的CEL文件或已经处理过的表达矩阵。为了方便后续分析,建议将数据转换为标准的表格格式,如CSV或TXT文件。
如果你使用的是R语言,可以使用以下代码来读取和预处理数据:
# 读取表达矩阵
expr_data <- read.csv("expression_matrix.csv", row.names = 1)
# 检查数据是否正确读取
head(expr_data)接下来,我们需要对数据进行标准化处理。标准化的目的是消除不同样本之间的系统性差异,使得各基因的表达水平具有可比性。常用的标准化方法包括Z-score标准化和log2转换。
# Z-score标准化
expr_data_scaled <- scale(expr_data)
# log2转换
expr_data_log2 <- log2(expr_data + 1)2. 构建加权共表达网络
在完成数据预处理后,我们可以开始构建加权共表达网络。WGCNA的核心思想是通过计算基因之间的相关性来构建网络。具体来说,我们可以通过Pearson相关系数或Spearman相关系数来衡量基因之间的相似性。
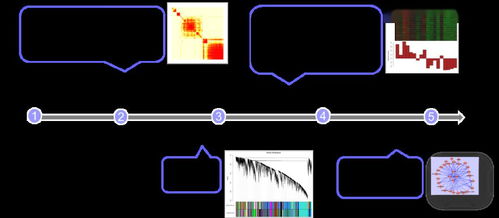
为了确保网络的稳定性,通常会选择一个合适的软阈值(soft threshold)。这个阈值的选择可以通过幂律分布图来确定。幂律分布图展示了不同阈值下网络的规模自由性(scale-free topology),选择一个使网络尽可能接近规模自由性的阈值是关键。
# 计算相关性矩阵
correlation_matrix <- cor(t(expr_data_scaled), method = "pearson")
# 选择软阈值
pickSoftThreshold(correlation_matrix, powerVector = c(1:20))根据幂律分布图的结果,假设我们选择了功率为6作为软阈值,那么接下来就可以构建加权邻接矩阵了。
# 构建加权邻接矩阵
adjacency_matrix <- adjacency(correlation_matrix, power = 6)3. 模块检测与可视化
构建好加权共表达网络后,下一步是进行模块检测。WGCNA通过层次聚类算法将基因分为不同的模块,每个模块内的基因具有较高的相关性。为了更好地理解这些模块的生物学意义,我们还可以将模块与临床特征或其他表型数据进行关联分析。
以下是模块检测的具体代码:
# 进行层次聚类
dissimilarity_matrix <- 1 - adjacency_matrix
gene_tree <- hclust(as.dist(dissimilarity_matrix), method = "average")
# 模块检测
modules <- cutreeDynamic(dendro = gene_tree, minClusterSize = 30)模块检测完成后,我们可以使用颜色编码的方式来可视化这些模块。这样不仅可以直观地展示模块的分布情况,还可以帮助我们发现一些潜在的生物学规律。
# 可视化模块
module_colors <- labels2colors(modules)
plotDendroAndColors(gene_tree, module_colors, "Module Colors", dendroLabels = FALSE, hang = 0.03, addGuide = TRUE, guideHang = 0.05)4. 模块-性状关系分析
最后,我们需要将模块与感兴趣的性状(如疾病状态、年龄、性别等)进行关联分析,以揭示模块与性状之间的关系。这一步骤可以帮助我们找到与特定性状密切相关的基因模块,从而为进一步的研究提供线索。
以下是模块-性状关系分析的具体代码:
# 读取性状数据
trait_data <- read.csv("trait_data.csv", row.names = 1)
# 计算模块-性状相关性
module_trait_cor <- cor(moduleEigengenes, trait_data, use = "p")
# 绘制热图
heatmap.2(module_trait_cor, col = colorRampPalette(c("blue", "white", "red"))(50), symm = TRUE, trace = "none", density.info = "none")5. 总结与展望
通过以上步骤,我们完成了一个完整的WGCNA分析流程。从数据准备到模块检测,再到模块-性状关系分析,每一步都至关重要。希望这篇文章能够帮助大家更好地理解和应用WGCNA技术,同时也欢迎大家在评论区分享自己的经验和问题。
在未来的学习中,我将继续深入探索WGCNA的高级功能,如多组学数据整合、动态网络分析等。相信随着技术的不断发展,WGCNA将会在生物医学研究中发挥越来越重要的作用。
如果你也对WGCNA感兴趣,不妨动手试试吧!期待与大家一起交流学习,共同进步。



发表评论 取消回复