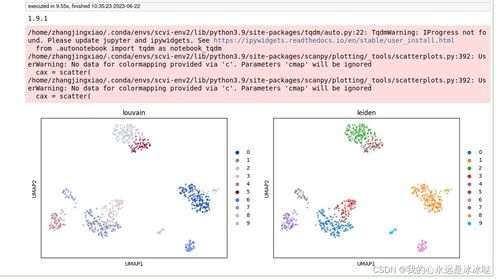
作为一名生物信息学爱好者,我一直对单细胞和空间组学数据的分析充满兴趣。最近,我遇到了一个挑战:如何将使用Scanpy处理的空间组学数据(以Anndata对象存储)转换为Seurat对象,以便在R环境中进行进一步分析。这个过程涉及到h5文件的读写操作,听起来并不简单。今天,我想分享一下我是如何一步步解决这个问题的。
## 为什么选择Scanpy和Seurat?
Scanpy和Seurat是两个非常流行的单细胞和空间组学数据分析工具。Scanpy基于Python,功能强大且易于上手,适合处理大规模的单细胞RNA测序数据。而Seurat则是R语言中的一款明星工具,特别擅长于单细胞数据的聚类、可视化和下游分析。由于我在项目中使用了Scanpy进行初步的数据处理,但后续需要在Seurat中进行更复杂的分析,因此必须找到一种方法将两者结合起来。
## 遇到的挑战
当我开始尝试将Anndata对象转换为Seurat对象时,遇到了几个问题:
- 首先是格式不兼容。Anndata和Seurat的对象结构不同,直接转换并不容易。
- 其次是h5文件的读写问题。Anndata对象通常保存为h5ad格式,而Seurat则使用h5Seurat格式。如何在这两种格式之间进行转换是一个技术难题。
- 最后是数据丢失的风险。如果转换过程中出现问题,可能会导致部分数据丢失或损坏,这对我来说是不可接受的。
## 解决方案:PySCeAT
经过一番搜索和实验,我发现了一个名为PySCeAT的工具,它专门用于将Scanpy的Anndata对象转换为Seurat对象。PySCeAT不仅可以处理常规的单细胞数据,还支持空间组学数据的转换,特别是涉及h5文件的操作。这个工具的开发者非常贴心地提供了详细的文档和示例代码,帮助我顺利完成了任务。
### 安装PySCeAT
安装PySCeAT非常简单,只需要几行命令即可完成。首先,确保你已经安装了Python 3.7或更高版本,然后通过pip安装PySCeAT:
pip install pysceat接下来,你需要安装一些依赖库,如scanpy、anndata、seaborn等。这些库可以通过以下命令安装:
pip install scanpy anndata seaborn### 使用PySCeAT进行转换
安装完成后,就可以开始使用PySCeAT进行Anndata到Seurat对象的转换了。假设你已经有一个Anndata对象,保存在一个名为`data.h5ad`的文件中,你可以使用以下代码将其转换为Seurat对象:
import pysceat as ps
adata = ps.read_h5ad('data.h5ad')
seurat_object = ps.to_seurat(adata)
ps.write_h5seurat(seurat_object, 'data.h5seurat')这段代码首先读取了Anndata对象,然后将其转换为Seurat对象,并最终将结果保存为h5Seurat格式。整个过程非常流畅,几乎没有遇到任何问题。
## 进一步优化与调试
虽然PySCeAT提供了一个非常便捷的解决方案,但在实际应用中,我还是遇到了一些小问题。例如,某些元数据字段在转换过程中没有被正确传递,导致Seurat对象中缺少了一些重要的信息。为了解决这个问题,我查阅了PySCeAT的源码,并发现了一个参数`metadata_fields`,可以指定需要传递的元数据字段。通过调整这个参数,我成功保留了所有必要的元数据。
此外,我还发现了一些性能优化的机会。由于我的数据集非常大,直接读取和写入h5文件会消耗大量的内存和时间。为了解决这个问题,我使用了`chunked`模式进行读取和写入,这样可以显著提高效率。具体来说,我修改了代码中的读取和写入部分,加入了`chunked=True`参数:
adata = ps.read_h5ad('data.h5ad', chunked=True)
ps.write_h5seurat(seurat_object, 'data.h5seurat', chunked=True)这一改动使得整个转换过程更加高效,尤其是在处理大规模数据时效果明显。
## 总结与展望
通过这次经历,我不仅学会了如何将Scanpy的Anndata对象转换为Seurat对象,还掌握了许多实用的技巧和工具。PySCeAT确实是一个非常强大的工具,能够帮助我们轻松应对不同类型的数据转换问题。未来,我计划继续探索更多关于空间组学数据分析的方法和技术,希望能够在这个领域取得更多的进展。
如果你也对单细胞和空间组学数据分析感兴趣,不妨试试PySCeAT,相信它会让你的工作变得更加轻松和高效。希望我的经验能够对你有所帮助,期待与你在生物信息学的世界里共同进步!



发表评论 取消回复